

2013年執業藥師藥理學知識點:喹諾酮類藥物概述
(一)簡史
萘啶酸(nalidixicacid)是1962年用于臨床的個喹諾酮類藥(實是萘啶酮),抗菌譜窄,口服吸收差,副作用多,現已不用。吡哌酸(pipemidicacid)抗菌活性強于萘啶酸,口服少量吸收,不良反應較萘啶酸少,可用于敏感菌的尿路感染與腸道感染。1979年合成諾氟沙星(norfloxacin),隨又合成一系列含氟的新喹諾酮類藥,通稱為氟喹諾酮類(fluoroquinolones)。
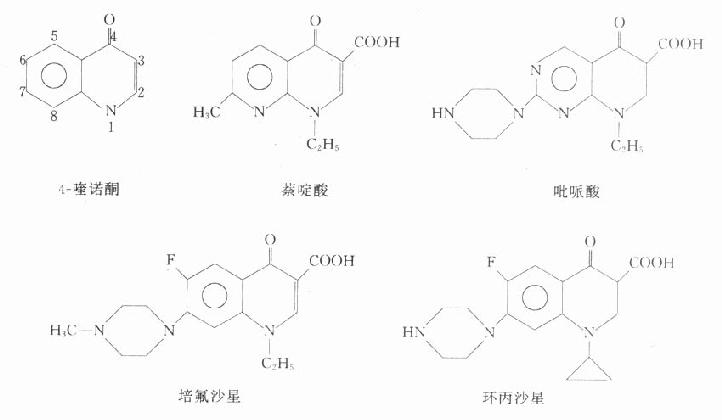
(二)化學結構與作用關系
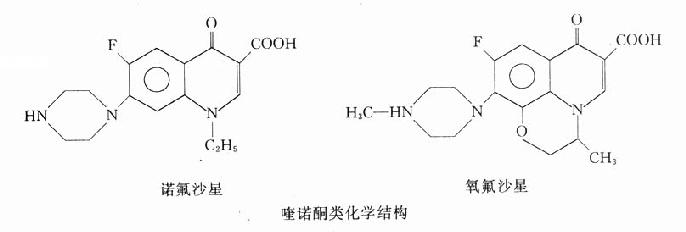
本類藥物的構效關系研究表明:4-喹諾酮母核的3位均有羧酸基,6位引入氟原子可增強抗菌作用并對金葡菌有抗菌活性;7位引進哌嗪環可提高對金葡菌及綠膿桿菌的抗菌作用(如諾氟沙星),哌嗪環被甲基哌嗪環取代(如培氟沙星),則脂溶性增加,腸道吸收增強,細胞的穿透性提高,半衰期延長。在8位引進第二個氟原子,可進一步提高腸道吸收,延長半衰期(如洛美沙星等),N-1修飾以環丙基團(環丙沙星)或噁嗪基團(氧氟沙星)可擴大抗菌譜,增強對衣原體、支原體及分支桿菌(結核桿菌與麻風桿菌等)的抗菌活性,噁嗪環還可提高水溶性,使藥物在體內不被代謝,以原形經尿排泄。
(三)抗菌作用機制
喹諾酮類通過抑制DNA螺旋酶作用,阻礙DNA合成而導致細菌死亡。大腸桿菌的DNA螺旋酶是四疊體結構的蛋白,由2個A亞單位與2個B亞單位組成,分子量分別為105kD與95kD(見圖42-1)。細菌在合成DNA過程中,DNA螺旋酶的A亞單位將染色體DNA正超螺旋的一條單鏈(后鏈)切開,接著B亞單位使DNA的前鏈后移,A亞單位再將切口封住,形成了負超螺旋。根據實驗研究,氟喹諾酮類藥并不是直接與DNA螺旋酶結合,而是與DNA雙鏈中非配對堿基結合,抑制DNA抑螺旋酶的A亞單位,使DNA超螺旋結構不能封口,這樣DNA單鏈暴露,導致mRNA與蛋白合成失控,后細菌死亡。
本類藥體外對DNA螺旋酶的半抑制濃度(IC50)與其對細菌的MIC呈一定的平行關系。哺乳動物的細胞內也含有生物活性與細菌DNA螺旋酶相似的酶,稱為拓樸異構酶Ⅱ(topoisomerase Ⅱ)。氟喹諾酮類藥對人體細胞拓樸異構酶Ⅱ影響較小(見表42-1)。從該表可見氧氟沙星與環丙沙星對動物細胞內拓樸異構酶Ⅱ的作用明顯比依諾沙星與萘啶酸小,IC50很高,選擇指數很大。這可能是氧氟沙星與環丙沙星不良反應較少的原因。
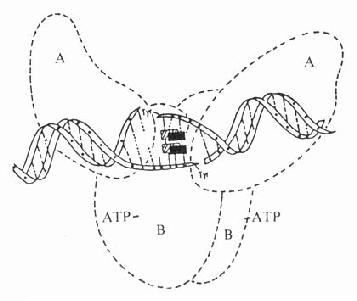
圖42-1 喹諾酮類藥物-DNA結合抑制DNA螺旋酶活性的示意圖
圖中實心和斜線長方形示喹諾酮類藥物分子,A、B為DNA螺旋酶的A、B亞單位。在DNA螺旋酶作用下,DNA雙鏈打開,而藥物分子嵌入雙鏈。與非配對堿基結合,阻礙DNA雙鏈封口
(四)細菌耐藥機制
氟喹諾酮類藥物廣泛應用后,已出現細菌耐藥性。耐藥機理研究證實主要是染色體突變,不存在質粒介導的耐藥性。耐藥機制有二:①細菌DNA螺旋酶的改變,與細菌高濃度耐藥有關;②細菌細胞膜孔蛋白通道的改變或缺失與低濃度耐藥有關。耐藥菌株DNA螺旋酶的活性改變主要由于gyrA基因突變所致。
(五)氟喹諾酮類藥理學共同特性
①抗菌譜廣,尤其對革蘭陰性桿菌包括綠膿桿菌在內有強大的殺菌作用,對金葡菌及產酶金葡菌也有良好抗菌作用;某些品種對結核桿菌,支原體,衣原體及厭氧菌也有作用;②細菌對本類藥與其他抗菌藥物間無交叉耐藥性;③口服吸收良好,部分品種可靜脈給藥;體內分布廣,組織體液濃度高,可達有效抑菌或殺菌水平;血漿半衰期相對較長,大多為3~7小時以上。血漿蛋白結合率低(14%~30%),多數經尿排泄,尿中濃度高;④適用于敏感病原菌所致的呼吸道感染、尿路感染、前列腺炎,淋病及革蘭陰性桿菌所致各種感染,骨、關節、皮膚軟組織感染;⑤不良反應少(5%~10%),大多輕微,常見的有惡心、嘔吐、食欲減退、皮疹、頭痛、眩暈。偶有抽搐精神癥狀,停藥可消退。
表42-1 喹諾酮類藥物對大腸桿菌和哺乳動物細胞DNA旋轉酶的選擇作用
| 藥物 | IC50(mg/L) | 選擇指數B/A | |
| A大腸桿菌KL-16DNA螺旋酶 | B胎牛胸腺局部拓樸異構酶Ⅱ | ||
| 氧氟沙星 | 0.76 | 1870 | 2461 |
| 環丙沙星 | 0.13 | 155 | 1192 |
| 依諾沙星 | 1.72 | 93 | 54 |
| 萘啶酸 | 23.0 | 385 | 17 |
相關推薦:
相關匯總:執業藥師藥學專業知識藥理學知識點匯總






